7.5. geom with xTB
As discussed in isomer with xTB, xTB is highly recommended for global optimization of chemical clusters that are unable to be described by force fields.
There are two ways of calling xTB in geom:
Use
misc/runxTB.shas discussed in isomer with xTB, just like
1commands
2 ./runxTB.sh $inp$ $out$ $xxx$
3end
To use this way, you must have an extra xTB installed on your system.
Use $xTB$ as discussed in Input File for geom.
1xtb
2 0 0 2
3end
4commands
5 $xTB$
6end
The advantage of this way is that you do NOT have to install xTB because these computation subroutines are already available internally in geom!
As discussed in Input File for geom, the three integers in xtb block are:
the charge of the whole cluster,
the number of unpaired electrons,
the Hamiltonian type (
0,1,2, or3(means GFN-FF)).
Tip
For atomic clusters, the Hamiltonian type is recommended to be set to 1. For other cases, 0, 1, and 2 are all good. For large organic or organometallic compounds, 3 may also be a good choice.
7.5.1. Example: NaCl(H2O)10
Tip
The sample input and output files can be found in testfiles/geom/4-naclh2o10-xTB.
In this Section we will see how to use geom to look for the global minimum of \(\text{NaCl}(\text{H}_2\text{O})_{10}\).
The structures of the 3 species are:
13
2water
3O 0.00000000 0.00000000 -0.11081188
4H 0.00000000 -0.78397589 0.44324751
5H -0.00000000 0.78397589 0.44324751
11
2Na
3Na 0 0 0
11
2Cl
3Cl 0 0 0
The input file is:
1lm_dir naclh2o10 # Save the local minima to this folder.
2num_calcs 2000 # Total number of calculations.
3do_coarse_opt yes # no: Do NOT the coarse optimization.
4min_energy_gap 1.E-4 # When two energies differ smaller than
5 # this value, they are treated as identical.
6 # A negative number means do not remove
7 # energetically degenerated ones.
8max_geom_iters 3000 # The maximum number of iterations for local optimization.
9 # If it is less or equal than zero, then the number is unlimited.
10
11components
12 h2o.xyz 10
13 random 0 0 0 8 8 8
14 ****
15 na.xyz 1
16 random 0 0 0 8 8 8
17 ****
18 cl.xyz 1
19 random 0 0 0 8 8 8
20 ****
21end
22
23xtb
24 0 0 1
25end
26
27commands
28 $xTB$
29end
You can see that, we let all molecules distribute randomly in a box defined by (0, 0, 0) and (8, 8, 8).
Now you can run the global optimization:
$ geom naclh2o10.inp > naclh2o10.out
After the optimization, the end of naclh2o10.out is
-- Result Report --
Results are energy-increasingly reordered.
Structures of energies within 1.000E-04 are treated as degenerate.
All minima are saved to "naclh2o10".
-------------------------------------------------------------------
# index Energy NaiveRMSD
-------------------------------------------------------------------
0 410 -78.90900677 0.00000000
1 1315 -73.85642855 11.13424019
2 270 -72.33437367 8.23789070
3 471 -71.89726049 2.84818350
4 595 -71.66037145 0.96099577
5 1138 -71.33515351 1.70326738
6 656 -71.30962706 6.91732743
7 309 -70.21312469 8.33181645
8 1007 -69.27883414 4.11576652
9 424 -68.96145859 1.36910299
10 777 -68.61390125 9.25461485
... omitted ...
28 966 -63.05453856 10.34934592
29 863 -62.49368470 9.79444040
30 1883 -62.49337541 9.23302757
31 648 -62.49194643 9.39246214
32 1811 -62.49123647 9.68901769
33 859 -62.49102429 9.53495472
Here you must be cautious. The lowest 29 structures, i.e. naclh2o10/410.xyz, naclh2o10/1315.xyz, … naclh2o10/966.xyz are highly dispersed, distorted unreasonable structures (see below), but they have uncommonly low energies. These misleading energies may occur when the initial guess is far away from equilibrium structures and often result from un-converged SCF processes.
After checking the energies, it seems that energies higher than -63 are normal ones. Indeed, the global minimum of this system is naclh2o10/863.xyz, which is shown below:

Tip
In practice, to further refine structures, like naclh2o10\863.xyz and naclh2o10/1883.xyz, you can use Gaussian or ORCA to do more accurate optimization.
7.5.2. Example: Co6Te8(PEt3)6
Tip
The sample input and output files can be found in testfiles/geom/5-co6te8pet36-xTB.
In this Section we will consider a quantum dot \(\text{Co}_6\text{Te}_8(\text{PEt}_3)_6\). It has a \(\text{Co}_6\text{Te}_8\) core ligated with 6 \(\text{PEt}_3\). Since we have no idea of its structure at this moment, we will search the core first.
7.5.2.1. Co6Te8
We can use exactly the same way as we have done for \(\text{NaCl}(\text{H}_2\text{O})_{10}\).
The structures of the 2 species are:
11
2Co
3Co 0 0 0
11
2Te
3Te 0 0 0
The input file is:
1lm_dir co6te8 # Save the local minima to this folder.
2num_calcs 2000 # Total number of calculations.
3do_coarse_opt yes # no: Do NOT the coarse optimization.
4min_energy_gap 1.E-4 # When two energies differ smaller than
5 # this value, they are treated as identical.
6 # A negative number means do not remove
7 # energetically degenerated ones.
8max_geom_iters 3000 # The maximum number of iterations for local optimization.
9 # If it is less or equal than zero, then the number is unlimited.
10
11components
12 co.xyz 6
13 random 0 0 0 8 8 8
14 ****
15 te.xyz 8
16 random 0 0 0 8 8 8
17 ****
18end
19
20xtb
21 0 0 1
22end
23
24commands
25 $xTB$
26end
Now you can run the global optimization:
$ geom co6te8.inp > co6te8.out
After the optimization, the end of co6te8.out is
-- Result Report --
Results are energy-increasingly reordered.
Structures of energies within 1.000E-04 are treated as degenerate.
All minima are saved to "co6te8".
-------------------------------------------------------------------
# index Energy NaiveRMSD
-------------------------------------------------------------------
0 954 -255485.30137014 0.00000000
1 1649 -13270.21996894 9.77918423
2 708 -379.80860329 8.25823482
... omitted ...
108 929 -51.90294094 9.92663264
109 29 -51.86444358 8.63886023
110 66 -51.86429930 9.29717029
111 199 -51.84758402 9.00426358
112 1213 -51.84370999 9.19951625
After checking the energies, reasonable ones should be higher than about -51.8. Thus, the global minimum should be co6te8/29.xyz, which is shown below.
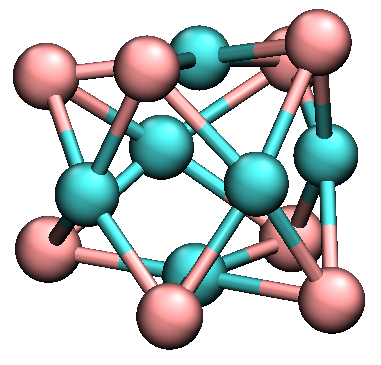
7.5.2.2. Co6Te8(PEt3)6
Now we will consider the ligation of \(\text{PEt}_3\), whose structure is:
1 22
2PEt3
3 P 0.77411167 0.93908628 0.00000000
4 C 1.38078883 1.79703460 -1.48602405
5 H 1.02413441 2.80584461 -1.48602405
6 H 2.45078883 1.79702166 -1.48602419
7 C 0.86744661 1.07107703 -2.74342827
8 H 1.19588317 1.59486033 -3.61674408
9 H 1.25178947 0.07263744 -2.76071851
10 H -0.20200553 1.04133445 -2.72647284
11 C 1.58017719 -0.68723832 0.13314147
12 H 1.06926441 -1.39066966 -0.49059924
13 H 2.59983880 -0.60671904 -0.18103498
14 C 1.53001448 -1.16683089 1.59569905
15 H 1.95386617 -2.14689639 1.66441129
16 H 2.08820972 -0.49286368 2.21139887
17 H 0.51311419 -1.19395588 1.92747918
18 C 1.16337141 1.93168334 1.47500099
19 H 0.62390995 1.54801668 2.31564434
20 H 0.88119749 2.94963161 1.30453342
21 C 2.67528550 1.85868155 1.75852616
22 H 3.05363867 2.84440501 1.93201431
23 H 2.84706388 1.25327881 2.62390451
24 H 3.17563928 1.42763883 0.91665428
For the core, we will just use the global minimum co6te8/29.xyz, which is renamed to co6te8.xyz and shown below:
114
2-51.86444358
3Co 5.18912366 4.53807660 5.15436968
4Co 4.63921329 6.73032694 4.33024999
5Co 5.57653575 4.28771551 2.73430585
6Co 3.73287281 2.69503667 3.34312049
7Co 3.09922668 5.44745620 2.69377997
8Co 2.32298551 4.63312799 4.83935341
9Te 3.65939955 5.91817891 6.45016152
10Te 5.16439545 6.53843136 1.92985727
11Te 1.55874524 3.57414579 2.71643092
12Te 3.68082433 2.75955616 5.79296888
13Te 5.94894157 2.39529023 4.27916498
14Te 2.22874466 7.03012888 4.38569672
15Te 3.80790879 3.72603676 1.12877717
16Te 6.88875884 5.89174763 3.98837449
The input file is:
1lm_dir co6te8pet36 # Save the local minima to this folder.
2num_calcs 200 # Total number of calculations.
3do_coarse_opt yes # no: Do NOT the coarse optimization.
4min_energy_gap 1.E-4 # When two energies differ smaller than
5 # this value, they are treated as identical.
6 # A negative number means do not remove
7 # energetically degenerated ones.
8max_geom_iters 3000 # The maximum number of iterations for local optimization.
9 # If it is less or equal than zero, then the number is unlimited.
10
11components
12 co6te8.xyz 1
13 fix 0 0 0 0 0 0
14 ****
15 pet3.xyz 6
16 shell 1 1 1 2 9 16 0 0 0 4 4 4
17 ****
18end
19
20xtb
21 0 0 1
22end
23
24commands
25 $xTB$
26end
From components, we can see that the core \(\text{Co}_6\text{Te}_8\) is fixed at (0, 0, 0) without rotation. Since its diameter is about 6 Å, we let the 6 \(\text{PEt}_{3}\) form a shell on an ellipsoid centered at (0, 0, 0) with radii 4, 4, and 4. They point to the shell with an axis defined by the average of atom 1, 1, 1 and 2, 9, 16.
Now you can run the global optimization:
$ geom co6te8pet36.inp > co6te8pet36.out
After the optimization, the end of co6te8pet36.out is
-- Result Report --
Results are energy-increasingly reordered.
Structures of energies within 1.000E-04 are treated as degenerate.
All minima are saved to "co6te8pet36".
-------------------------------------------------------------------
# index Energy NaiveRMSD
-------------------------------------------------------------------
0 5 -192.73555542 0.00000000
1 69 -192.73510648 5.73710764
2 87 -192.73423439 5.54349689
3 107 -192.73409359 4.88639799
4 197 -192.73377634 5.63049822
5 136 -192.73339919 5.57130216
6 89 -192.73317855 5.26383292
So the global minimum is co6te8pet36/5.xyz, which is shown below.
