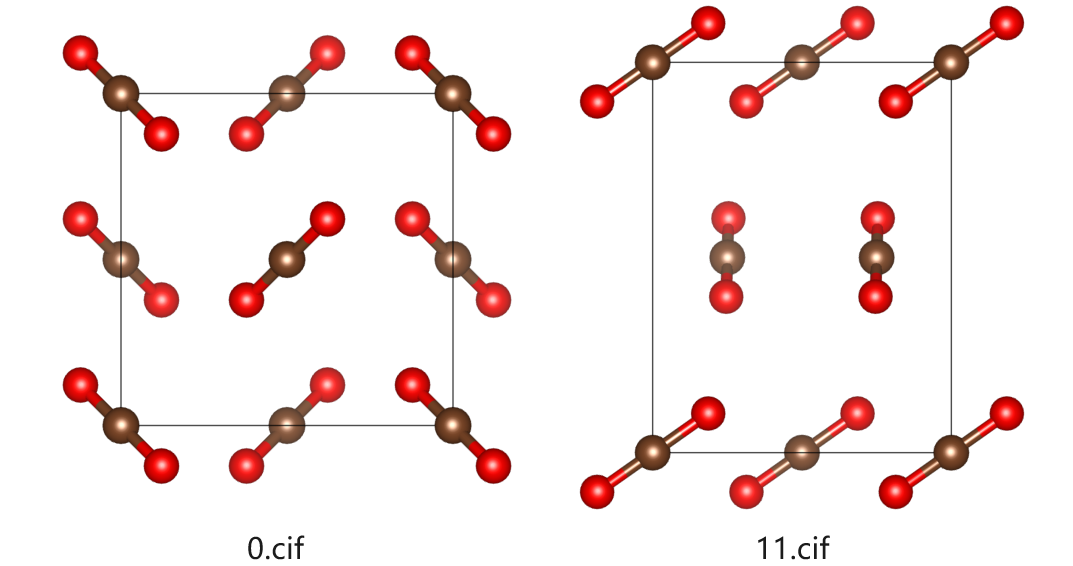
3.2. Example: CO2 Crystal
Tip
The sample input and output files can be found in testfiles/1-co2.
In this first example, we will try to predict the crystal structure of \(\text{CO}_2\).
3.2.1. Run the Search
Before proceeding, we should have a parameter file for \(\text{CO}_2\) molecule and an initial guess of crystal shape.
For the parameter file, you can check if it has already be provided in
mics/charmm36. In this case, yes: you can find a filemics/charmm36/co2.xyz. That is. In not, you have to build the paramter by yourself. This will be introduced Example: Build Parameter File for (NHCH3)2CO.In addition, you need an initial guess for crystal shape: the number of molecules in a cell (\(Z\)) and the crystal system. If experimental values are known, we can just use them as input. Otherwise, we have to try different values.
Step 1: Prepare an input file named sys.inp with the following content (other file names are also acceptable):
1sys # Result file name
2sys.crystal # Crystal components file name
334 # Crystal family: 31-37
420 # ABC argument: Population
55 # ABC argument: Maximum generation
65 # ABC argument: Scout limit
71000 # Number of local minima to be saved
83 # Cutoff of the neighboring cells. For non-polar: 3; polar or charged: 4-6
91.0 # Structures with intramolecular atoms smaller than this distance will be discarded.
100.1 # Structures with density smaller than this value will be discarded.
110.02 # When two energies differ smaller than this value, they (of the same symmetry) are treated as identical.
The texts after # are comments and can be arbitrary. The input will be explained line by line:
The folder name to save predicted crystal structures.
The name of the file that contains crystal components, cell initial guess and optionally geometrical information. It will be explained in the next step.
An integer representing the crystal system that will be kept during the search. The following input are acceptable:
Input |
Unit |
|---|---|
|
triclinic |
|
monoclinic |
|
orthorhombic |
|
tetragonal |
|
trigonal |
|
hexagonal |
|
cubic |
One can consider more than cyrstal systems, for example 35 37 stands for trigonal and cubic lattice. Note that, during optimization, the symmetry may raise (say, triclinic cell may become a cubic one), but will rarely be lowered.
The population size \(SN\). See Theoretical Background for details.
The maximum cycle number \(g_{\mathrm{max}}\). See Theoretical Background for details.
The scout limit \(g_{\mathrm{limit}}\). See Theoretical Background for details.
The number of crystal structures to be saved. During the calculations, there may be a large number of generated structures. This is a upper limit of the structures to be saved.
This is a cutoff parameters for short-ranged force evaluations. Usually,
3is suffucient. If higher accuracy is needed, a larger value like4can be set. However, larger values will lead to more expensive energy evaluation.This is a distance in Angstrom. A structure with intramolecular atoms smaller than this distance will be discarded. Usually,
1.0is a good choice.This is a density in g/cm 3. A structure with density samller than this value will be discarded. For example, if you know (maybe from experiments) that the crystal density is around 2 g/cm:sup:3, then you can set this value to
1.7. If you are not sure, simply set it to0.1.This is an energy difference in kJ/mol. Two structures with energy difference smaller than this value will be discarded. Usually,
0.02is a good choice.
Step 2: Copy misc/charmm36/co2.xyz to the current path. Then prepare the cluster file named sys.crystal with the following content:
11 1.01325
2co2.xyz 4
36 6 6 90 90 90
4*
The first number is kind of molecules in the crystal. The second number is the pressure in bar. The ambient pressure is
1.01325bar.The molecule parameter file name, and its number (\(Z\)).
Tip
In the crystal file, the parameter files can be given with relative or absolute path. For example, if the water parameter file is at /home/you/abcrystal/misc/tip4p.xyz and you do not want to copy it, you can simply give the absolute path of this file in sys.cluster like /home/you/abcrystal/misc/tip4p.xyz.
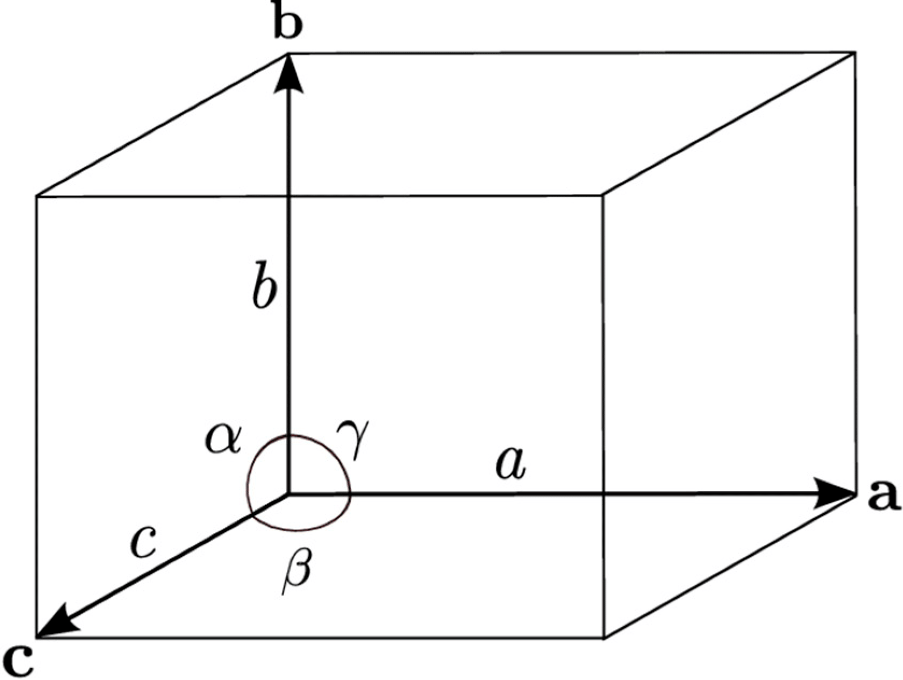
The initial guess of crystal shape: \(a,b,c,\alpha,\beta,\gamma\) (see the Figure below). Note that, the number does NOT have to meet the crystal system constraint. The symmetrization will be treated by ABCrystal.
A
*is needed. Note that,abcrystalwill also generate*.crystalfiles, but it does not end with*but with some real numbers, for example:
11 1.01325000
2co2.xyz 4
3 5.53512686 5.53522121 6.00000000 90.00000000 90.00000000 90.00000000
4Energy: -107.74033984
5 3.93647424 0.95531967 2.35617388 0.00000000 0.00000000 0.00000000
6 4.68931899 0.95532737 5.49777255 0.49999755 0.00000000 0.49999760
7 1.68906741 5.32785973 0.78540539 0.00000052 0.49999714 0.49999743
8 3.38675967 0.95532317 0.78541716 0.49999807 0.49999755 0.00000019
These numbers are rigid coordinates of each molecule in the crystal. You do not need to care about them.
Step 3: Run the crystal structure prediction:
$ abcrystal sys.inp > sys.out
After a few seconds, you will find several new files:
sys.outThe main output file.sys-OPT.crystalThe most stable crystal structure in ABCrystal format.sys-OPT.xyzThe most stable crystal structure in XYZ format.sys-OPT.gjfThe most stable crystal structure in GJF format.sys-OPT.cifThe most stable crystal structure in CIF format.sysA folder containing all crystal strucures, each one having 4 files in ABCrystal, XYZ, GJF, and CIF format, respectively. They are sorted in energy-increasing order, e.g.0.xyzis lower in energy than13.xyz.abcrystal*.crystal/xyz/gjf/cifThe file containing the currently found most stable sturcture during the running ofabcrystal. You can check the current stable structure beforeabcrystalterminates. If it crashes, one can use thisabcrystal*.crystalto start a new search.
3.2.2. Check the Output
We can now open sys.out. You can go to Optimization Results to check the may results:
1 # Energy Vcell Density Space ID Bravais Good?
2 (kJ/mol) (AA^3) (g/cm^3) Group Lattice
3 0 -107.7403 169.5847 1.7237 Pa-3 205 cP Y
4 1 -107.6989 169.5989 1.7235 Pa-3 205 cP Y
5 2 -107.6493 169.6153 1.7233 Pa-3 205 cP Y
6 3 -107.4886 169.6683 1.7228 Pa-3 205 cP Y
7 4 -106.8056 169.9081 1.7204 Pa-3 205 cP Y
8 5 -106.3587 170.0616 1.7188 Pa-3 205 cP Y
9 6 -104.1541 168.7669 1.7320 Pca2_1 29 oP Y
10 7 -103.3376 170.5019 1.7144 Pnnm 58 oP Y
11 8 -103.3130 170.5116 1.7143 Pnnm 58 oP Y
12 9 -102.5955 170.8538 1.7109 Pnnm 58 oP Y
1310 -96.5469 168.6160 1.7336 Pbca 61 oP Y
1411 -94.2686 177.0464 1.6510 P2_1/c 14 mP Y
1512 -94.2302 177.0638 1.6508 P2_1/c 14 mP Y
1613 -94.0207 177.1590 1.6500 P2_1/c 14 mP Y
1714 -92.9351 167.4234 1.7459 P-1 2 aP Y
1815 -92.4825 175.7740 1.6630 P2_1/c 14 mP Y
Let’s see line 3:
0 -107.7403 169.5847 1.7237 Pa-3 205 cP Y
The 1st index means that this structure corresponds to file
sys/0.crystal/xyz/gjf/cif.The 2nd number is the enthalpy:
-107.7403kJ/mol.The 3rd number is the cell volume:
169.5847Angstrom 3.The 4th number is the density:
1.7237g/cm 3.The 5th string is the space group name international symbol:
Pa-3, i.e., \(\text{Pa}\bar{3}\).The 6th number is the space group ID:
205.The 7th string is the Bravais lattice symbol:
cP(primitive cubic lattice).
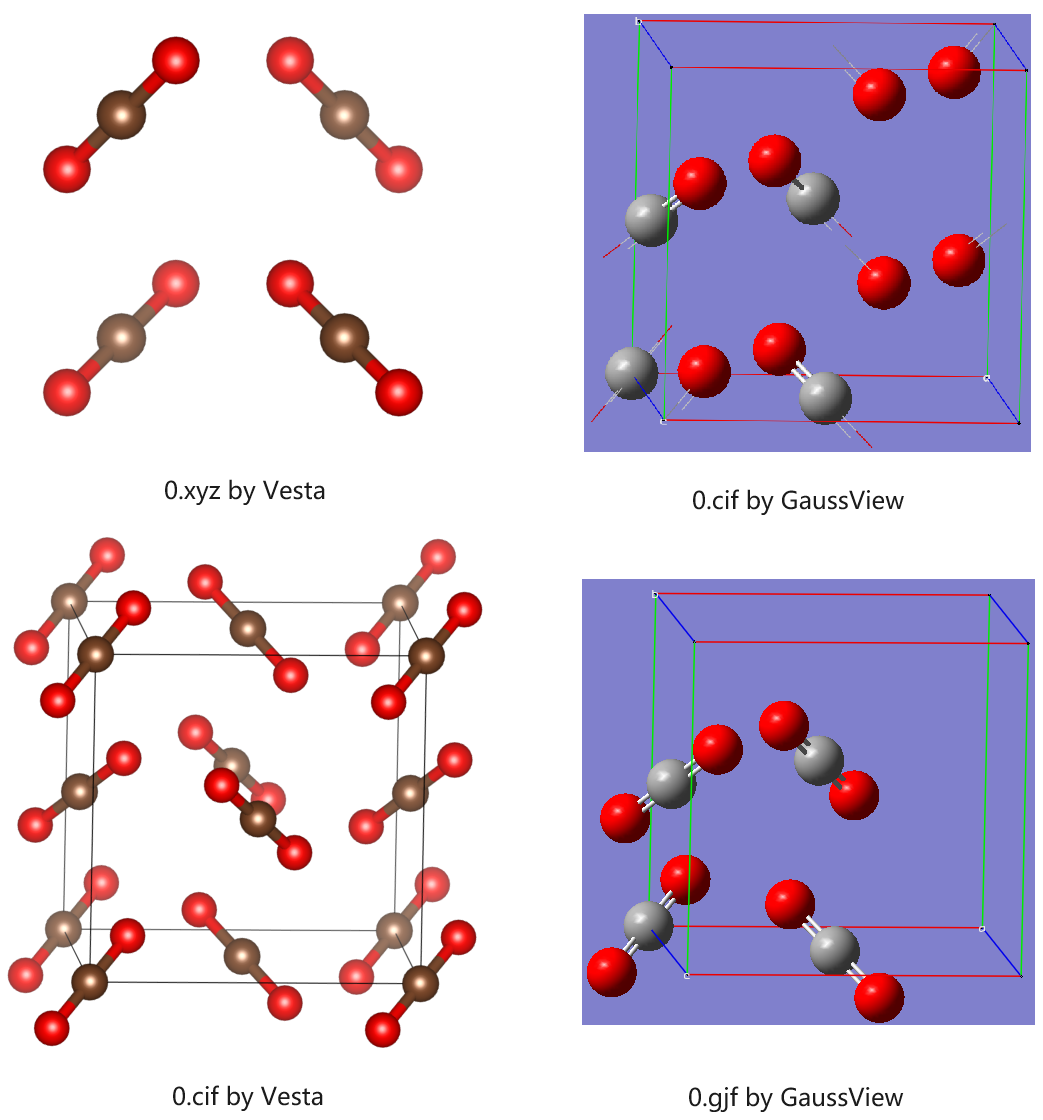
3.2.3. Visualization of Structures
The structures can be visualized in different ways.
XYZ file does not contain cell information. It only contains Cartesian coordinates and can be used in, e.g., molecular dynamics.
GIF file contains both atomic and cell information and can be visualized with GaussView. All molecules can be shown in a wrapped way.
CIF file is the standard crystal file. If can be viewed with Vesta and GaussView, but molecules may not be wrapped.
Below is a summary of visualization of structure sys/0.xyz/gjf/cif.
3.2.4. DFT Calculations
The energy in ABCrystal is caluclated with CHARMM force field. A better energy estimation way is to use density functional theory. Here, we will use CP2K to do a PBE-D3 calculation for 2 crystal structures.
Copy
misc/crystal-cp2k-dft.inpto a suitable path, say,cp2k-calcs, and change the name to0.inpand11.inpWe want to see structure
0and11energy difference. Copysys/0.cifandsys/11.ciftocp2k-calcs.Open
0.inp, and change@SET sysnameline to
@SET sysname 0
In
&SUBSYS ... &END SUBSYS, you cna add corresponding basis set and pseudopotential. In&DFT ... &END DFT, you can change the calculation method. Here, nothing needs to be changed.In
&CELL_OPT ... &END CELL_OPT, you can change pressure, symmtery constraint, etc. according to your need.After editting
0.inp, run the calculation:
cp2k -i 0.inp -o 0.out
Run the calculation for
11in a similar way.
When the calculations are finished, the optimized atomic position is in 0/11-pos-1.xyz, and cell information is in 0/11.out. We can compare their results:
Method |
CHARMM |
PBE-D3 |
|---|---|---|
E(11)-E(0) (kJ/mol) |
13.47 |
15.91 |
Cell volume 0 (Angstrom 3) |
169.58 |
164.33 |
Cell volume 11 (Angstrom 3) |
177.05 |
178.39 |
The structures are shown below: